INTRODUCTION
In modern society, individuals are prone to lacking vitamin D as they are exposed to situations that reduces vitamin D activations or supplementation such as in spending more time indoors and the scarcity of food sources that are high in the vitamin containment. As a result, vitamin D insufficiency is emerging as an important public health challenge. A study finds that possibly up to 40% of Europeans are deficient in vitamin D level [1]. Based on a cutoff value of serum level 25(OH)DŌēż20 ng/mL, nearly 1 billion individuals worldwide would exhibit vitamin D insufficiency.
Following a study in which intake of 100 IU of vitamin D raised serum levels by around 0.7ŌĆō1 ng/mL, the treatment guidelines of The United States Endocrine Society recommend that adults aged Ōēź19 years should take at least 1,500ŌĆō2,000 IU of vitamin D each day to maintain an optimal concentration of 30 ng/mL [2,3]. Generally, in order to raise serum 25(OH)D by 10 ng/mL, an individual needs to take 1,000 IU/day of vitamin D3 for 3ŌĆō4 months [3].
Vitamin D3 B.O.N. Intramuscular Injection (cholecalciferol) is a drug recommended for supplementation of vitamin D3 developed in France in 1964. It has been used for over 50 years worldwide and has been demonstrated to be safe and effective.
Prior studies investigated efficacy and safety of either a single high-dose injection of vitamin D or multiple injections to the very elderly but there have been no studies that looked over efficacy and safety of injection of vitamin D3 over a long span of time. Thus, in attempts to find out the longevity in efficacy and safety in injection supplementation of the vitamin, we conducted an investigator-initiated clinical trial and examined the efficacy and safety of regular intramuscular injections of vitamin D3 B.O.N.
We aim to evaluate efficacy and safety of regular administration of high dose intramuscular injections of vitamin D3 and establish guidelines for the regular administration of the drug.
METHODS
Participants
The study included adults aged 19ŌĆō65 years with vitamin D deficiency who showed serum 25(OH)D<20 ng/mL on the screening day, and who had voluntarily signed the participant consent form after understanding the study content and goals. The recruitment of the participants was conducted openly at Soonchunhyang University Seoul Hospital.
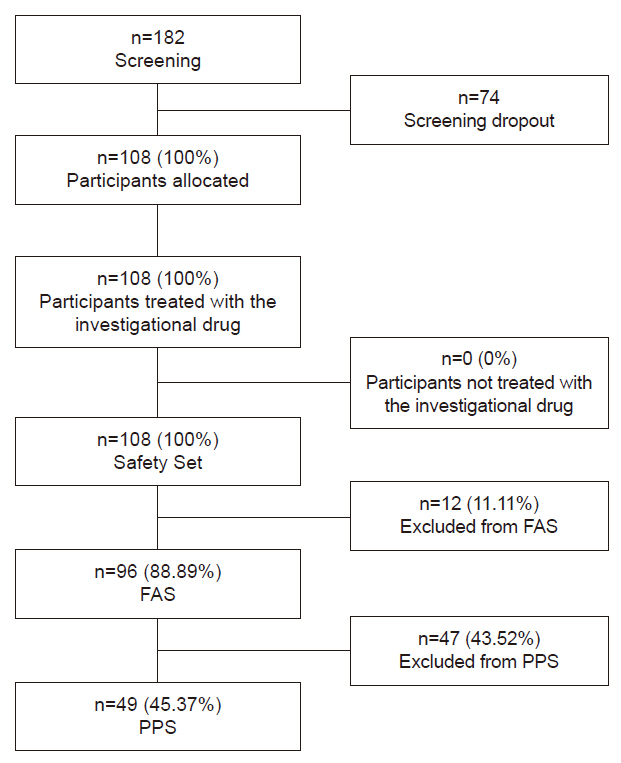
The final participants included in the analysis consisted of 108 persons, after 74 people were screened out of 182 originally recruited, prior to commencement of trials, due to reasons such as testing above or equal to level of 20 ng/mL of serum 25(OH)D, self-cancelations of consents by the participants, concurrently participating in other clinical trial, inability to meet the planned schedules, missing of consensual authorization, and belonging to vulnerable group, in the order of prevalence of the incidents.
Of the 108 participants included, there were 59 cases that were dropped out during their trials due to reasons such as follow up loss (52 cases), self-cancelations of consents by the participants (three cases), decisions made by principal investigator to cease trial for safety (2:1 case of pregnancy, one case categorized vulnerable group), decision made by principal investigator to cease trial arbitrarily (one case of unrelated incident that caused study drug interruption) and concurrently participating in other clinical trial (one case), in the order of prevalence of the incidents (Fig. 1).
Approval and review
This study of the vitamin D clinical trial was reviewed and approved by the Institutional Review Board at Soonchunhyang University Seoul Hospital (KDBON-CPI-01). This clinical trial complied with the principles of the International Conference on Harmonization (ICH) Guidelines and the Declaration of Helsinki and was conducted in accordance with the related regulations in consideration of subject rights and safety. Written informed consent was obtained from all subjects.
The investigational drug
Study process
The study was conducted over a period of 22 months (observation period per participant: approximately 12 months). The investigational drug (vitamin D3 B.O.N. Intramuscular Injection [cholecalciferol]) was administered via intramuscular injection at a dose of 200,000 IU in the first visit and the subjects were followed up at 3-month intervals to be checked for serum 25(OH)D levels along with other indicators including physical examinations, vital signs, serum levels of 1,25(OH)2D, and calcium concentration, urine levels of calcium and creatinine and other concurrent medications. The drug was re-administered in the subjects if serum 25(OH)D was tested to show below the normal range (30 ng/mL) during the follow up visits.
As an investigator-initiated clinical trial to examine the efficacy and safety of the drug, endpoints were set in primary and secondary efficacy, and safety. The efficacy endpoints were defined as the mean changes in serum levels of 25(OH)D (primary) and 1,25(OH)2D (secondary), each from their baselines to 3 months, from baselines to 6 months, from baselines to 9 months, and from baselines to 12 months. The percentage of participants that resulted within the normal ranges of serum 25(OH)D level (30ŌĆō100 ng/mL) at 3, 6, 9, and 12 months were investigated as secondary efficacy endpoint as well. Safety endpoints consisted of physical examination, vital signs, adverse events (AEs), blood calcium concentration, and urine calcium and creatinine concentrations.
Analyses
The data obtained from the subjects of this clinical trial were analyzed in the form of FAS (Full Analysis Set), PPS (Per Protocol Set), and Safety Set. The FAS included all data obtained from subjects who were registered as trial subjects, received the investigational drug after enrollment, and had at least one primary efficacy evaluation performed after baseline. The per-protocol analysis group (PPS) was set apart within the FAS set according to the protocol compliance, given the nature of the open-label trial. The compliance refers to the minimum treatment duration, usability of measurements, and the absence of major protocol violations. PPS group included only those subjects who sufficiently adhered to the clinical trial protocol. As this clinical trial involved subjects who could have prior knowledge of the administered drug in an open-label trial, the conventional intention-to-treat principle for general randomization was not applied. The Safety Set included all data obtained from all trial subjects who received the investigational drug at least once.
In order to test the efficacy of the investigational drug for blood 25(OH)D and 1,25(OH)2D, we presented descriptive statistics for each treatment group to analyze the changes in concentration at each time point relative to baseline. We analyzed the change at each time point using either two sample t-test or WilcoxonŌĆÖs rank sum test, depending on whether or not the assumption of normality was satisfied. The percentage of participants within the normal serum 25(OH)D range (30ŌĆō100 ng/mL) at 3, 6, 9, and 12 months after the first dose was shown as the count and percentage of participants, and analyzed using PearsonŌĆÖs chi-square test or FisherŌĆÖs exact test.
In order to test safety, we presented the descriptive statistics for each treatment group for AEs occurring after exposure to the investigational drug. We analyzed AEs using PearsonŌĆÖs chi-square test or FisherŌĆÖs exact test. In addition, descriptive statistics were presented for AEs after drug administration, adverse drug reactions (ADRs), and severe adverse events (SAEs). In terms of measurements and test values, for continuous variables (physical examination, vital signs, and blood calcium concentration), descriptive statistics were presented for the baseline values, values at trial termination, and the differences between baseline and termination. The variables were analyzed using paired t-test or WilcoxonŌĆÖs signed rank test. For categorical variables, contingency tables were constructed and the variables were analyzed using McNemarŌĆÖs test.
The results of analysis were presented in terms of odds ratios and 95% confidence interval. All data analyzes were performed using SAS ver. 9.4 (SAS Institute Inc.) with a significance level of 5%.
RESULTS
ParticipantsŌĆÖ general characteristics
Ninety-six participants analysed included 23 males (23.96%) and 73 females (76.04%), with a mean age of 47.46┬▒12.98 years, and a mean height of 161.02┬▒7.61 cm. The detailed results are presented in Table 1.
Primary efficacy endpoint
For the 96 participants included in the FAS group, the mean change in blood 25(OH)D from baseline to 3, 6, 9, and 12 months was 12.89┬▒8.93 ng/mL, 17.96┬▒8.51 ng/mL, 15.77┬▒8.26 ng/mL, and 17.13┬▒8.46 ng/mL, respectively. The changes after treatment with the investigational drug compared with baseline were statistically significant at all time points (all P<0.0001). The PPS group of 49 participants showed similar trends compared with the FAS group, with statistically significant changes at all time points after exposure to the investigational drug compared with baseline (all P<0.0001). The detailed results for blood 25(OH)D levels are shown in Table 2.
Secondary efficacy endpoint
The mean change in blood 1,25(OH)2D from baseline to 3, 6, 9, and 12 months in the 96 participants in the FAS group, was 11.36┬▒16.42 pg/mL, 17.33┬▒20.34 pg/mL, 20.64┬▒21.41 pg/mL, and 19.07┬▒21.65 pg/mL, respectively. The changes after administering the investigational drug compared with baseline were statistically significant at all time points (all P<0.0001). The PPS and FAS groups showed similar trends, with statistically significant changes at all time points after treatment with the investigational drug compared with baseline (all P<0.0001). The results of blood 1,25(OH)2D levels are shown in Table 3.
The percentage of participants remaining in the normal serum 25(OH)D level (30ŌĆō100 ng/mL) after the injection was 19 (19.79%) at 3 months after baseline, 43 (44.79%) at 6 months, 36 (37.50%) at 9 months, and 40 (41.67%) at 12 months respectively out of the 96 participants in the FAS group. The 49 participants of PPS group showed 11 (22.45%) at 3 months after baseline, 28 (57.14%) at 6 months, 20 (40.82%) at 9 months, and 26 (53.06%) at 12 months, respectively, showing higher percentage at all time points compared to that of the FAS group. The analysis is summarized in Table 4.
Safety assessment
AEs were analysed in the group of Safety Set using data collected since immediately after the informed consent (screening) until the participantŌĆÖs final assessment visit (the final visit in the 12 months period or by dropout).
Of the 108 participants in the Safety Set, 14 (12.96%) experienced AEs, and four (3.70%) experienced suspected ADRs. No SAEs or severe ADRs occurred during the study period. Two of the 16 cases of AEs (12.50%) exhibited ongoing symptoms, but they were both deemed unrelated to the investigational drug. The other 14 AEs (87.50%) were resolved, and no SAEs were detected during the study period.
No statistically significant changes in blood/urine calcium and creatinine concentrations were found at the end of the trial compared with baseline, and no clinically significant abnormal results were detected at any time point.
All criteria of discernment of SAEs were based on the guidelines set out by International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), which defines a serious AE (experience) or reaction as ŌĆ£any untoward medical occurrence that at any dose: results in death, is life-threatening, requires inpatient hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability/incapacity, or is a congenital anomaly/birth defect.ŌĆØ
Details of the AEs are listed in Table 5.
Safety assessment details
A total of 16 AEs were detected in 14 participants. These included four ADRs, with three participants experiencing a single painful event at the injection site, and a participant reporting an incident of injection site pruritus. In all, the symptoms were mild and transient and all patients recovered without further treatment.
In terms of events affecting treatment with the investigational drug, a single ŌĆśstudy drug interruptedŌĆÖ case (6.25%) was observed in relation to urinary calculus. However, the event was assessed not to be related to the drug. The patient was recommended extracorporeal shockwave lithotripsy, and based on the investigatorŌĆÖs judgment, the patient was removed from the trial without further follow-up.
Two ongoing (12.50%) AEs, assessed as unrelated to the investigational drug, regarded a participant suffering a lower limb fracture and a participant developing urinary calculus. They were found to be ongoing after receiving concomitant medication or non-drug therapy.
When all the AEs after drug administration were classified by organ, there were three cases of injection site pain (2.78%) and one case of injection site pruritus (0.93%). However, these were confirmed to be transient AEs caused by the injection formulation.
In addition, there were three cases of myalgia (2.78%), two cases of dizziness (1.85%), and one case each of fatigue (0.93%) and urinary calculus (0.93%), but these were not predictable AEs based on the articles of approval for the studied drug.
There was one case each of herpes zoster (0.93%), nasopharyngitis (0.93%), sinusitis (0.93%), gastritis (0.93%), and lower limb fracture (0.93%), but none of these were on going or showed any suspected association with the injection, and were resolved after appropriate treatment.
In physical examination, no participants showed clinically significant abnormalities after the injection. No statistically significant changes in systolic blood pressure were detected at the end of the trial compared with baseline. Statistically significant decreases in diastolic blood pressure, pulse rate, and body temperature at were found at the end of the trial compared with baseline, but these were not clinically significant.
DISCUSSION
There have been numerous studies that showed correlations of risks of different diseases to lower levels of serum vitamin D levels. Studies found that vitamin D deficiency increases the risk of falls and fractures in falls, as well as autoimmune disease, cardiovascular disease, infectious disease, colorectal cancer, and type 2 diabetes [4]. Individuals with low vitamin D levels or genetic variants in vitamin D receptor were reported to show higher incidence of malignant tumors such as breast cancer, colorectal cancer, and prostate cancer [5-8], hypertension [9], diabetes [10], and diseases associated with immune disorders. However, a contradicting study is also seldomly found such as in the findings, through meta-analyses of 15 randomized controlled trials, where preventive measure from high-dose supplementation of vitamin D were minimal on risk of falls and fractures [11]. They further speculated that it may even increase the risk.
Despite the disputed ideas, ample evidence suggests that vitamin D deficiency is strongly associated with multiple skeletal and non-skeletal diseases and laboratory and epidemiological studies to date indicate that appropriate vitamin D levels are essential to maintaining optimal health.
Although currently there is no consistent criteria for the determination of appropriate serum 25(OH)D concentrations, a serum level of 25(OH)D<10 ng/mL is generally defined as ŌĆśdeficientŌĆÖ, 10ŌĆō30 ng/mL as ŌĆśinsufficientŌĆÖ, and Ōēź30 ng/mL as ŌĆśsufficientŌĆÖ. The World Health Organization defines <10 ng/mL as ŌĆśdeficientŌĆÖ and Ōēż20 ng/mL as ŌĆśinsufficientŌĆÖ [12].
Different academic groups suggest differing levels as being appropriate levels of serum vitamin D, such as National Academy of Medicine formerly known as the Institute of Medicine suggesting 20 ng/mL or above, yet a study by Bischoff-Ferrari et al. [13] defines appropriate serum 25(OH)D as Ōēź30 ng/mL. In another study, Bischoff-Ferrari et al. [14] suggest the appropriate 25(OH)D concentration recommended is 30ŌĆō44 ng/mL. Precise guidelines for treating vitamin D deficiency have yet to be defined, but usually individuals with a blood vitamin D (25[OH]D) concentration <20 ng/mL are considered for treatment.
Currently, vitamin D formulations include oral formulations and injections. Based on the results of multiple studies, a daily vitamin D intake of 800ŌĆō1,000 IU is recommended to prevent vitamin D insufficiency [15]. However, treating deficiency requires supplementation of a higher dose of vitamin D, which would be achieved more effectively through injections.
This study result enhances the evidence, priorly proven through studies of a single high dose injection of vitamin D in elderly or multiple injections in the regard [15], that injection is an effective and safe method for supplementation of vitamin D, and further provides information that the efficacy and safety remain valid relatively over a long period. This observance may be beneficial to people who require or prefer injections over other methods of supplementations.
The mainstay of the study was to observe efficacy and safety of the injection by seeing the serum 25(OH)D level within normal ranges, and regular repeated injections with its potential to overload the level was not considered in this study. The overlooked investigations for the number of the participants who required additional shots, the number of the shots each received, and their base serum level of 25(OH)D would have better elaborated the investigated drugŌĆÖs capacity to enhance the serum concentration.
The percentage of participants in the normal range (30ŌĆō100 ng/mL) for blood 25(OH)D after the injection was higher in the PPS group at all time points compared to that of the FAS group, supporting the grounds, by the definition of each group, that participants who followed the protocol more strictly also more effectively gained and maintained improvements. But this can also implicate people who generally adhere better to healthy guidelines, and having better overall exposure to vitamin D, followed the protocol better.
Trials were initialized on participants regardless of the time within a year and seasonal effects on serum 25(OH)D level, such as from differing sun light exposure, might have been dismissed.
Even though vitamin D is known to promote calcium absorption in the small intestine and long-term high-dose vitamin D administration had been reported to have caused hypercalcemia or renal calculi [15], the lack of clinically significant abnormal results measured in blood calcium concentration and in blood/urine calcium and creatinine concentrations at any time point, along with that of any other findings in physical examination, vital signs, or AEs demonstrate the safety of the injection.
Conclusion
This clinical trial shows that treatment of vitamin D-deficient adults with B.O.N. Intramuscular Injection 200,000 IU induces a statistically significant increase in blood vitamin D concentration demonstrating the clinical efficacy of the intervention. Further, the higher percentage of the group that complied better to the guidance being within targeted normal ranges strengthens the evidence of the efficacy the drug holds. No clinically significant new AEs or SAEs were associated with the injections demonstrating its safety. Thus, administration of vitamin D3 B.O.N. Intramuscular Injection is safe and effective in vitamin D-deficient adults aged 19ŌĆō65 years.
Further studies that demonstrate the role of outdoor activity and occupational categories, use of ultraviolet blockers, different age and sex groups will be desirable. Such studies may facilitate development of appropriate guidelines for interventions using vitamin D3 intramuscular injections.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print